PHANS-C Tutorial¶
For those who want to hit the ground running…
You can either use a pre-made Amazon AWS AMI, Docker image, or install PHANS-C locally.
For tips getting PHANS-C running, and getting the tutorial folder, check out Get PHANS-C.
Get the tutorial folder¶
- AWS users will find the folder in
/home/ubuntu/Tutorial - Docker users will need to download the Tutorial folder, then mount it as a shared folder (see Get PHANS-C)
- Local users will find the tutorial folder in the main PHANS-C repository download.
The Tutorial¶
The order of the tutorial steps roughly follow the flow diagram of the pipeline found here. Open the Tutorial directory. From here on out, paths are relative to the Tutorial directory.:
If you prefer, there is a shell script which automates the following process. However, we recommend that you manually run through the steps to gain a greater understanding. If you prefer to use the shell script for the tutorial data, it can be run as follows:
$ sh run_tutorial.sh
Warning: If you installed PHANS-C and prerequisites manually, rather than using the AWS image, it is very common to have to slightly modify some scripts to get the workflow running. For example, the raxml binary can be called different things (e.g. raxml, raxmlHPC, raxmlHPC-AVX, raxml-AVX2, raxml_8.2.11, etc) depending on how it was installed. You may need to open the run_tutorial.sh and ProteinModelSelection.pl scripts and adjust the name of the raxml binary. Be on the lookout for other similar errors. Another example is the third party ProteinModelSelection.pl script may need to have the perl path modified on the first line of the script.
1: Construct the Phylogenetic Reference Tree¶
First, you will need to assemble a list of full length gene sequences which will act as known reference sequence which will be used to compare the unknown metagenomic sequences. For this tutorial, we will identify and categorize sequences of the ribosomal protein rpsD. Full length rpsD reference sequences in amino acid space can be found in the file AA/rpsD_AA.fasta.
1.1: Make reference sequence alignment¶
Move into the AA directory:
$ cd AA/
Generate an alignment for the reference rpsD sequences using the program muscle.
$ muscle -in rpsD_AA.fasta -out rpsD_AA_aln.fasta
1.2: Convert alignment to phymlAln format¶
Convert from fasta to phymlAln format for tree construction.
$ phan-fastaToPhyml.py -f rpsD_AA_aln.fasta
phan-fastaToPhyml.py command line options:
| -h, --help | Show this help message and exit. |
| -f FILE | Specify the FASTA in-file that contains the sequence alignment. |
1.3: Find best amino acid substitution model for reference tree¶
Since we are using amino acid space, you may want to test for the best amino acid substitution model. However, you may use any model available in RAxML. An excellent perl script, proteinModelSelection.pl, for identifying the best AA substitution model for tree generation was created by the RAxML author Alexis Stamatakis and can be found at the RAxML site.
The proteinModelSelect.pl script generates a lot of output files,so, to manage these create a new directory:
$ mkdir ProteinModel
Will need to move a copy of the alignment file into this directory in order to run proteinModelSelection.pl.:
$ cp rpsD_AA_aln.phymlAln ./ProteinModel/
Now, move into the ProteinModel directory.
$ cd ProteinModel
Run proteinModelSelection.pl and redirect the standard output, which is a printout of the best substitution model, to the file BestModel.txt.
$ proteinModelSelection.pl rpsD_AA_aln.phymlAln > BestModel.txt
When proteinModelSelection.pl is finished running, check for the best identified substitution model.
$ cat BestModel.txt
Which should print out Best Model: LG
Also, check to see if a file rpsD_AA_aln.phymlAln.reduced has been generated. If there are duplicate sequences, this .reduced file is generated, and it is best to construct a tree on this alignment file. You can find what sequences were duplicates and which were removed from the alignment by checking the RAxML_info files.
$ more RAxML_info.LG_rpsD_AA_aln.phymlAln_EVAL
Move a copy of the .reduced file back to the AA directory, which should be one level above the current directory.
$ cp rpsD_AA_aln.phymlAln.reduced ../
Now, move back to the AA directory.
$ cd ../
1.4: Construct an amino acid reference phylogenetic tree¶
Build a tree using the generated alignment files and the best amino acid subtiution model identified. You can find more information about constructing trees with RAxML at the RAxML software page.
The PHANS-C Pipeline data analysis was developed using RAxML version 8.2.8. On your system, the RAxML exectuable may be named differently. For example, on our local system, ours is called raxmlHPC-PTHREADS-AVX. On the AWS AMI, version 8.2.8 can be accessed via the AMI executable raxml; however, a newer version of RAxML, version 8.2.11, is also available on the AMI and has been tested for compatibility with the PHANS-C Pipeline. RAxML version 8.2.11 can be accessed through the AMI executable raxml_8.2.11 or via raxml.
$ raxml -m PROTCATLG -s rpsD_AA_aln.phymlAln.reduced -# 1 -T 8 -p 610 -n rpsD_AA
Some of the options used to construct this tree are as follows:
| -m STR | Designates the model used to build the tree. Here, we are constructing an amino acid tree PROT using a gamma model of rate heterogeneity GAMMA using the best amino acid substitution model printed by proteinModelSelection.pl which is LG… PROTGAMMALG |
| -s FILE | Specify the input alignment file. |
| -T INT | Specify the number of threads. Do not use more than your computer has available. |
| -p INT | Specify a random seed for starting parsimony inferences. |
| -n STR | Specify a title for the output file names. |
-# INT Specify the number of distinct starting trees.
For expediency in running this tutorial, we are using the CAT model and only using 1 starting tree.However, we would strongly encourage more stringent settings for real data, and recommend using the GAMMA model of rate heterogeneity -m PROTGAMMALG and at least 10 starting trees -# 10.
See the RAxML documentation for further explanation of options.
2: Parse and Format reads from Metagenome¶
2.1: Construct a BLAST database from the metagenomic sequence reads¶
You will need BLAST+ in order to build a blast database. You can find information on obtaining BLAST Get PHANS-C. The BLAST database will need to be hash indexed in order to pull metagenomic reads from the database for placement.
First, move into the blast directory:
$ cd ../blastDB/artificialMG/
An artificial metagenome, artificialMG.fasta, is located in this directory. Now, use this file to generate a blast database.
$ makeblastdb -parse_seqids -hash_index -dbtype nucl -in artificialMG.fasta -out artificialMG
In order to access this blast database from any directory, we will need to add it to the blast path. If you have it installed locally, use your own custom location. If you are using the AWS image, to add this directory to the blast path for this session of the shell, enter the following in the command line:
$ export BLASTDB=$HOME/Tutorial/blastDB/artificialMG
In order to add this directory to the blast path for every session, update the shell .profile with the following:
$ echo 'export BLASTDB=$HOME/Tutorial/blastDB/artificialMG/' >> ~/.profile
2.2: Search the BLAST database for potential reads of interest¶
Use BLAST search to pull out reads from the metagenome which show similarity to your target gene of interest. In this case, parse out reads that show similarity to a set of rpsD sequences we are interested in. We will use a set of full length amino acid sequences as queries from the file ~/Tutorial/AA/rpsD_AA_seeds_for_blast.fasta. NOTE: It is important to use full length gene sequences, as the trimming script used later will trim the metagenomic reads to be within the length or your starting query sequences.
Move to the directory AA
$ cd ../../AA/
Now use this amino acid sequence seed file to search a 6-frame translation of the metagenomic reads.
$ tblastn -db_gencode 11 -db ../blastDB/artificialMG/artificialMG -outfmt 5 -num_threads 2 -max_target_seqs 1000000 -evalue 1 -query rpsD_AA_seeds_for_blast.fasta -out rpsD_artificialMG.tblastn.xml
2.3: Parse reads from the BLAST results¶
Take the outfile from this BLAST search, rpsD_artificialMG.tblastn.xml, and parse out the desired information:
$ phan-parseBlastXML.py -f rpsD_artificialMG.tblastn.xml -n 0 -e ../blastDB/artificialMG/artificialMG -s -l -o rpsD_artificialMG_blastResults -c 2
phan-parseBlastXML.py command line options:
| -h, --help | Show this help message and exit. |
| -f FILE | Specify the blast XML format FILE that you wish to use as input. |
| -o FILE | Specify the outfile that you wish to use. If nothing is specified, the name and location of the infile will be used, with a “best” as the suffix. |
| -n INT | Specify the number of hits you want from each blast record. A 0 means return all results. Default is 1. |
| -s, --sort | Sort by evalue. |
| -e BLASTDB | Specify db to extract hit read sequences from. |
| -c INT | Specify number of cpu cores to use. Default=2. |
phan-parseBlastXML.py output files:
rpsD_artificialMG_blastResults.hits- A file of all the blast hits: contains the blast information as well as the sequence of the reads. Blast results partitioned by query sequence. This file to be passed as input to next pipeline stepEach hit has a fasta title in the following tab delimited format: Query Hit Accession Description Score Evalue Hit Number Query Length Hit ID Hit Length Align Length Identities Positives Gaps Query From Query To Hit From Hit To Frame Query Frame Hit ##### #Hits below from query Acaryochloris_marina_MBIC11017_66 ##### >SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 No definition line 130.0 3.53312e-10 1 202 SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 142 42 25 30 0 89 130 1 126 0 -2 TCGGTAGCTGGGAATATCCACGCGCTTGCCGTTGACCAGGATGTGACCGTGACCCACAAACTGCCGGGCCTGACGGCGGGTCGAGGCAAACCCCATGCGGAACACGACATTGTCGAGGCGGCGCTCCAGCAGCTGCAGGAAC >SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 No definition line 130.0 3.53312e-10 2 202 SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 142 42 25 30 0 89 130 1 126 0 -2 ...
rpsD_artificialMG_blastResults- A tab delimited file of all the unique blast hits with associated blast result information.:Query Hit Accession Description Score Evalue Hit Number Query Length Hit ID Hit L ength Align Length Identities Positives Gaps Query From Query To Hit F rom Hit To Frame Query Frame Hit Acaryochloris_marina_MBIC11017_66 SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_3197 95_37835_2 SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 No definition line 130.0 3.53312e-10 1 202 SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonom yId_319795_37835_2 142 42 25 30 0 89 130 1 126 0 -2 Acaryochloris_marina_MBIC11017_66 SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_3197 95_37835_2 SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 No definition line 130.0 3.53312e-10 2 202 SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonom yId_319795_37835_2 142 42 25 30 0 89 130 1 126 0 -2 ...
rpsD_artificialMG_blastResults.hits_all.fasta- A file that just contains all of the read sequences for the blast hits.:>lcl|SMPLB_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_2 TCGGTAGCTGGGAATATCCACGCGCTTGCCGTTGACCAGGATGTGACCGTGACCCACAAACTGCCGGGCCTGACGGCGGG TCGAGGCAAACCCCATGCGGAACACGACATTGTCGAGGCGGCGCTCCAGCAGCTGCAGGAAC >lcl|SMPLB_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_58376_2 TTTTTACCGTTAACTCTTACGTGACCATGGTTAATAAGTTGTCTCGCTGAAAATACTGTTGTTGAAAATCTTGCTCTATA AACAACAGAATCTAATCTTCTTTCAAGCAAACCAATTAAATTTTCACCCGTATCCCCTTTTAACAT ...
2.4: Trim the metagenomic reads:¶
Pass the .hits output file from the previous step to phan-trimFormatReads.py in order to format and clean the reads for placement.:
$ phan-trimFormatReads.py -g rpsD -f rpsD_artificialMG_blastResults.hits -m 100 -e 1e-5
phan-trimFormatReads.py command line options:
| --version | Show program’s version number and exit. |
| -h, --help | Show this help message and exit. |
| -g STR | Specify the gene name which will be used to label output files. |
| -f FILE | Specify the input file that is parsed from blast output… *.hits file. |
| -m INT | Specify the minimum length of reads that will be passed on to rest of analysis, if no min specified all reads will be passed. |
| -e STR | (OPTIONAL) Evalue cutoff all reads must be =< this evalue to continue, if no max specified all reads will be considered. Enter in scientific notation (1e-5). |
| -s INT | (OPTIONAL) All reads must be >= this score to continue, if no min specified all reads will be considered. |
Functions of phan-trimFormatReads.py:
- Trims the reads so that they are in the proper reading frame which corresponds with the BLAST hit.
- Trims the reads to remove any overhang that might exist - ie. if the read captures the beginning of a gene, but also sequence upstream, the upstream sequence will be removed to improve alignment quality.
- Sets an e-value or score threshold for reads - must meet certain similarity scores in order to continue for placement analysis.
- Sets a length requirement - reads, after trimming, must be of a specific length (designated in nucleotide bases) in order to continue for placement analysis.
- Removes any reads with ambiguous nucleotide bases - creates a file with all the reads which ambiguous nucleotides.
- Removes stop codons in the amino acid translation (the stop codons do not work with downstream placement software). List of reads with stop codons produced.
phan-trimFormatReads.py output files:
rpsD_formatted_query_seqs_NT.fasta- A fasta file of the trimmed read sequences. Trimmed according to the blast hit information.:>SMPLA_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_58376_1 AAAGTTAACATTGGAAGTTACGTTGTTAAAGAAGAAGATATAATTGAAATTAGAGATAAATCTAAACAGTTGGCTATAATTGATATCGCTCTAGCTAGTAA AGAAAGAGAAACACCTGAATACATTAATTTAGATGAAAAAAAT >SMPLA_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_58376_2 ATGTTAAAAGGGGATACGGGTGAAAATTTAATTGGTTTGCTTGAAAGAAGATTAGATTCTGTTGTTTATAGAGCAAGATTTTCAACAACAGTATTTTCAGC GAGACAACTTATTAACCATGGTCACGTAAGAGTTAACGGTAAA >SMPLB_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_79864_1 AAACATAAAATAGATAGAAGGTTAAAAATTAACCTATGGGGTAGACCTAAAAGTCCTTTTAATAAAAGGGATTATGGTCCAGGACAACACGGACAAGGTAG AAAAGGAAAACCTTCTGATTACGGAATTCAGCTACAAGCTAAACAAAAATTAAAA ...
rpsD_AA_translation_stops_removed.fasta- A fasta file of the amino acid translation of the trimmed sequence reads according to the blast hit information. Any stop codons (*) have been removed as stop codons interfere with downstream programs. A file calledrpsD_AA_translations_with_Stops.fastais also produced which contains any reads with stop codons.:>SMPLA_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_58376_1 KVNIGSYVVKEEDIIEIRDKSKQLAIIDIALASKERETPEYINLDEKN >SMPLA_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_58376_2 MLKGDTGENLIGLLERRLDSVVYRARFSTTVFSARQLINHGHVRVNGK >SMPLB_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_79864_1 KHKIDRRLKINLWGRPKSPFNKRDYGPGQHGQGRKGKPSDYGIQLQAKQKLK ...
rpsD_info_DICTIONARY_FILE_for_back_translate.csv- A data file in .csv format which contains key sequence information.:Accession, e-value, abs_frame, raw_read, fixed_read, AA_with_stop, AA_removed_stop >SMPLA_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_58376_1, 1.15292e-24, 3, ATTTTTTTCATCTAAATTAATGTATTCAGGTGTTTCTCTTTCTTTACTAGCTAGAGCGATATCAATTATAGCCAACTGTTTAGATTTATCTCTAATTTCAATTATATCTTCTTCTTTAACAACGTAACTTCCAATGTTAACTTTTT, AAAGTTAACATTGGAAGTTACGTTGTTAAAGAAGAAGATATAATTGAAATTAGAGATAAATCTAAACAGTTGGCTATAATTGATATCGCTCTAGCTAGTAAAGAAAGAGAAACACCTGAATACATTAATTTAGATGAAAAAAAT, 233.0, KVNIGSYVVKEEDIIEIRDKSKQLAIIDIALASKERETPEYINLDEKN, KVNIGSYVVKEEDIIEIRDKSKQLAIIDIALASKERETPEYINLDEKN ...
rpsD_unique_reads_with_ambiguous_nucleotides.fasta- If there are reads which have ambiguous nucleotides (e.g. base = ‘N’), then those reads are removed and listed here.
rpsD_query_reads_processing_info.log- A helpful basic stats file that should be reviewed before proceeding to next step of the pipeline. If there are significant reads which are removed due to ambiguous nucleotides or a high abundance of reads with stop codons, then the user should proceed with caution.:The original number of blast reads prior to deduplication = 3110 The original number of unique reads recruited by blast = 106 e-value cutoff of 1e-05 used; all reads reported are <= this e-value threshold. Number of reads which did not make this e-value threshold and were discarded: 23 The number of unique reads recruited from blast that also met thresholds specified = 83 The number of unique reads containing ambiguous nucleotides = 0 The percent of unique reads with ambiguous nucleotides that were removed from analysis = 0.00% The number of unique reads that were less than 100 bases & removed from analysis = 7 The percent of reads that were too short & removed from analysis = 8.43% The number of clean unique reads without ambiguous nucleotides = 76 The number of reads with at least one stop codon = 0 Percent of unique reads with stop codons = 0.00%
3: Align unknown reads to Reference Alignment¶
3.1: Use the program PaPaRa to align reads¶
Combine the formatted unknown reads with the reference phylogeny for identification. Take the alignmment that was used to construct the phylogentic reference tree and align all the unknown clean reads (amino acid space) to the reference alignment using the software package PaPaRa.
$ papara -t RAxML_bestTree.rpsD_AA -s rpsD_AA_aln.phymlAln.reduced -q rpsD_AA_translation_stops_removed.fasta -n rpsD_AA_artificialMG -j 1 -a -f
PaPaRa command line options used:
| -t FILE | Specify the reference phylogenetic tree. |
| -s FILE | Specify the alignment used to build the reference tree. |
| -q FILE | Specify the file with the formatted environmental sequence reads. |
| -n STR | Specify label for output files. |
| -j INT | Specify number of threads (do not specify more threads than available on your machine). |
| -a | Using amino acid sequences. |
PaPaRa output files:
papara_alignment.rpsD_AA_artificialMG - The alignment file in phymlAln format which contains the recruited reads aligned to the reference alignment.
140 228
Bifidobacterium_angulatum_DSM_20098_21 --MTNVQRSRRQVRLSRALGIAL----------TPKAQRIFEKRPYAPGEHG--RTRRRTESDYAVRLREKQRLRAQY-GLSEKQLRAVYEKGTKTSGQTGNAMLQDLEVRLDNLVLRAGFARTTAQARQFVVHRHILVDGNIVNRPSYRVKPGQTIQVKAKSQTMEPFQAAAEGVHRDVLPPVPGYLDVNLASLKATLTRKPEAEE--V-PAQVNIQYVVEFYAR--
Pirellula_sp_1_61 MKVTMARYTGPKARINRRLGTMLYET-----AGAARA---MDRRPQPPGMHT----RGRRPSNYGAALMEKQKIKHYY-GLGERQLRRYFENVGRKSGNTGELLLLMCERRLDNVVRRVGFTKTRPQARQGITHGHFRVNGVKVTKPGYMLRAGDLIEVRGRENLKNLYRGVIANSPPDGLD----WVSFDSETLRATVLSLPGAVD--I-SLPVDANSVVEFLSR--
Planctomyces_brasiliensis_DSM_5305_53 ----MGRYTGPKARINRRLGSPVFES-----AGALRA---SEKRDSPPGMH---QRRK-KPTIYGAALTEKQKIKYYY-GFRERQLRKYFNEARRLKGNTGQNLLVLCERRLDNVVRRAGFAQTRPQARQAIVHSHFQLNGRTVNKPSIQVRAGDVITVRNRPNLHAIYKEQVGRADTAACE----FVSVDDSALKIVVSAIPTFAD--V-SLPVDVNQVVAFLSR--
...
SMPLB_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_32557_2 -------------------------------------------------------------------------------------------------------------------------------------------------------KEEDIIEIRDKSKQLAIIDIALA-SKERETPE---YINLDEKNKKVTFVRTPKFDE---------------------
...
papara_log.rpsD_AA_artificialMG - Settings and statistics for the PaPaRa run. This information is also printed to StdOut upon running PaPaRa.
papara called as:
papara -t RAxML_bestTree.rpsD_AA -s rpsD_AA_aln.phymlAln.reduced -q rpsD_AA_translation_stops_removed
.fasta -n rpsD_AA_artificialMG -j 16 -a -f
references container instantiated as: papara::references<pvec_pgap, sequence_model::tag_aa>
edges: 125
scoring scheme: -3 -1 2 -3
papara_core version 2.4
start scoring, using 16 threads
thread 1: 0.786583 gncup/s
thread 3: 0.378382 gncup/s
thread 10: 0.200862 gncup/s
thread 6: 0.37806 gncup/s
thread 4: 0.371692 gncup/s
thread 9: 0.193493 gncup/s
thread 8: 0.378985 gncup/s
thread 12: 0.209433 gncup/s
thread 2: 0.477034 gncup/s
thread 13: 0.177332 gncup/s
thread 14: 0.282384 gncup/s
thread 0: 0.286568 gncup/s
thread 15: 0.283097 gncup/s
thread 11: 0.327415 gncup/s
thread 5: 0.594323 gncup/s
thread 7: 0.489285 gncup/s
scoring finished: 0.023149
generating best scoring alignments
SUCCESS 0.147885
3.2: Join reads to increase phylogenetic signal¶
Next generation sequencing technologies often use mate pairing or paired ends - geneating 2 reads per strand of genomic DNA. In order to enhance the phylogenetic signal of the unknown reads for placement, and reduce errors of duplicate counting, the script spaceJoin will combine the alignments of individual read pairs into one read for placement. Since the exact length between the read pairs is unknown, an assumption is made that the alignment of the individual reads is accurrate. Read pairs should be designated as _1 and _2 in fasta file used for building the BLAST database, and the optional suffix _0 can represent read pairs that have already been joined.
$ phan-spaceJoin.py -p papara_alignment.rpsD_AA_artificialMG -r 64 -o rpsD_AA_cut40 -l 40
phan-spaceJoin.py command line options:
| --version | Show program’s version number and exit. |
| -h, --help | Show this help message and exit. |
| -p FILE | Specify the output file from PaPaRa that has the reference aln and metagenome reads aln combined (“papara_alignment.*”). |
| -r INT | Specify the number of sequences in the reference alignment (# of sequences in reference tree). |
| -o FILE | Specify the your desired output alignmemt file name. |
| -l INT | Specify the minimum length of each read for placement; default set to 45 bases/residues. |
phan-spaceJoin.py output files:
Log_spaceJoin_rpsD_AA_cut40_joinedReadsForPlacement.txt- A log file that outputs statistics about the joining of read pairs. Reads that are joined are relabeles as “_3”.The following of the input alignment are the following: Pre-Joind BOTH READS: 0 BOTH READS: 29 ONLY READ 1: 12 ONLY READ 2: 6 ***************************************************************************************** The following are the set of reads in the output file that are longer than 40 LONG Pre-Joined BOTH READS: 0 LONG BOTH READS: 29 LONG ONLY READ 1: 12 LONG ONLY READ 2: 4 Perecent of reads from total dataset that are longer than 40: 97.37% Percent of long reads with pairs: 78.38% ***************************************************************************************** Reads that are written to output file Reads with mate pairs joined: SMPLA_Vibrio_sp_MED222_NCBI_taxonomyId_314290_82469_3, SMPLB_Vibrio_sp_MED222_NCBI_taxonomyId_314290_82469_3, SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_3, SMPLA_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_19173_3, ...
rpsD_AA_cut40_joinedReadsForPlacement.phymlAln- A new alignment file with the joined reads.109 228 Bifidobacterium_angulatum_DSM_20098_21 --MTNVQRSRRQVRLSRALGIAL----------TPKAQRIFEKRPYAPGEHG--RTRRRTESDYAVRLREKQRLRAQY-GLSEKQLRAVYEKGTKTSGQTGNAMLQDLEVRLDNLVLRAGFARTTAQARQFVVHRHILVDGNIVNRPSYRVKPGQTIQVKAKSQTMEPFQAAAEGVHRDVLPPVPGYLDVNLASLKATLTRKPEAEE--V-PAQVNIQYVVEFYAR-- Pirellula_sp_1_61 MKVTMARYTGPKARINRRLGTMLYET-----AGAARA---MDRRPQPPGMHT----RGRRPSNYGAALMEKQKIKHYY-GLGERQLRRYFENVGRKSGNTGELLLLMCERRLDNVVRRVGFTKTRPQARQGITHGHFRVNGVKVTKPGYMLRAGDLIEVRGRENLKNLYRGVIANSPPDGLD----WVSFDSETLRATVLSLPGAVD--I-SLPVDANSVVEFLSR-- ... SMPLA_Escherichia_coli_W3110_NCBI_taxonomyId_316407_83762_3 -------------------------------------------------------------------------------------FRNYYKEAARLKGNTGENLLALLEGRLDNVVYRMGFGATRAEARQLVSHKAIMVNGRVVNIASYQVSPNDVVSIREKAKKQSRVKAALELAEQREKPT--------------------------------------------- SMPLB_Candidatus_Pelagibacter_ubique_HTCC1062_NCBI_taxonomyId_335992_8052_3 ----MTKRISAKHKIDRRLKINL--------WGRPKS--PFNKRDYGPGQHG--QGRKGKPSDYGIQLQAKQKLKGYYGNINERQFRNIYKKAT-------------------------------------------------------------------------------------------------------------------------------------- ...
4: Phylogenetic placement of unknown reads¶
4.1: Use the Evolutionary Placement Algorithm (EPA) to assign unknown reads within tree¶
The formatted unknown metagenomic reads are combined with the reference phylogeny using RAxML EPA.
$ raxml -f v -m PROTCATLG -s rpsD_AA_cut40_joinedReadsForPlacement.phymlAln -r RAxML_bestTree.rpsD_AA -n rpsD_AA_placements -T 8
RAxML-EPA command line options used:
| -f v | Use EPA to place environmental sequence reads onto a reference tree. |
| -m PROTGAMMALG | Use the PROTGAMMALG model. |
| -s FILE | Alignment file with joined environmental reads aligned to reference alignment. |
| -r FILE | Specify the original reference tree. |
| -n STR | Label for output files. |
| -T INT | Specify number of threads (do not specify more threads than available on your machine). |
RAxML-EPA output files:
RAxML-EPA produces numerous files - details of which can be found in the RAxML manual.
RAxML_classification.rpsD_AA_placements - Read placement/classification results file.
SMPLB_Bacteroides_plebeius_DSM_17135_NCBI_taxonomyId_484018_1483_1 I9 1 0.00000100000050002909
SMPLB_Shewanella_baltica_OS155_NCBI_taxonomyId_325240_2956_1 I96 1 0.00000100000050002909
SMPLA_Bacteroides_plebeius_DSM_17135_NCBI_taxonomyId_484018_1483_1 I9 1 0.00000100000050002909
SMPLA_Shewanella_baltica_OS155_NCBI_taxonomyId_325240_2956_1 I96 1 0.00000100000050002909
...
RAxML_classificationLikelihoodWeights.rpsD_AA_placements - Read placement/classification results using likelihood weights.
RAxML_entropy.rpsD_AA_placements - Entropy results using likelihood weights.
RAxML_originalLabelledTree.rpsD_AA_placements - Original reference tree with labeled node IDs.
RAxML_labelledTree.rpsD_AA_placements - Reference tree with branch labels and read placements.
RAxML_portableTree.rpsD_AA_placements.jplace - Labelled reference tree with branch labels in portable pplacer/EPA format (without query sequences). This file can be used along with the package genesis for viewing/manipulating placement data. genesis is not included within the AWS AMI or the PHANS-C Pipeline.
4.2: Visualize distribution of likelihood scores (or branch-lengths)¶
All of the reads may not be placed with high quality scores. In order to evaluate read placement quality, you can view an interactive visualization of the distribution of either likelihood scores for the final placement of a read or the branch lengths. The script “phan-distributions.py” will generate a stand-alone interactive html file where you can inspect the distribution of reads (see figure below for example). Below the chart of the distribution is a table of the reads sorted in order of the score bins. By clicking on a bar in the chart, it will highlight the corresponding row in the table. You can then use keyboard shortcuts to copy the row of values (the reads with scores in that bin) and paste them into a text editor.
The following will generate a distribution of the likelihood scores in a stand-alone .html file:
$ phan-distributions.py -i RAxML_classificationLikelihoodWeights.rpsD_AA_placements -l -o rpsD_likelihoods
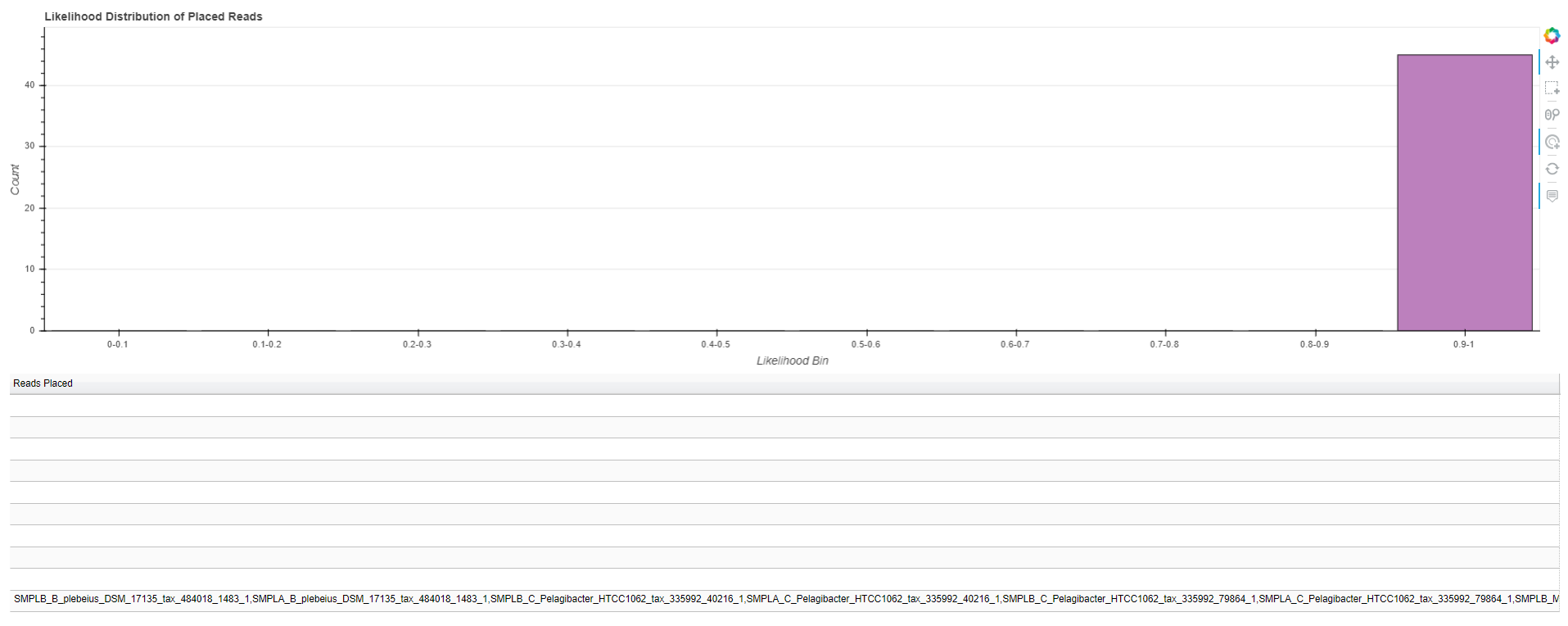
The following will generate a distribution of branch lengths in a stand-alone .html file:
$ phan-distributions.py -i RAxML_classification.rpsD_AA_placements -b -o rpsD_branchLengths
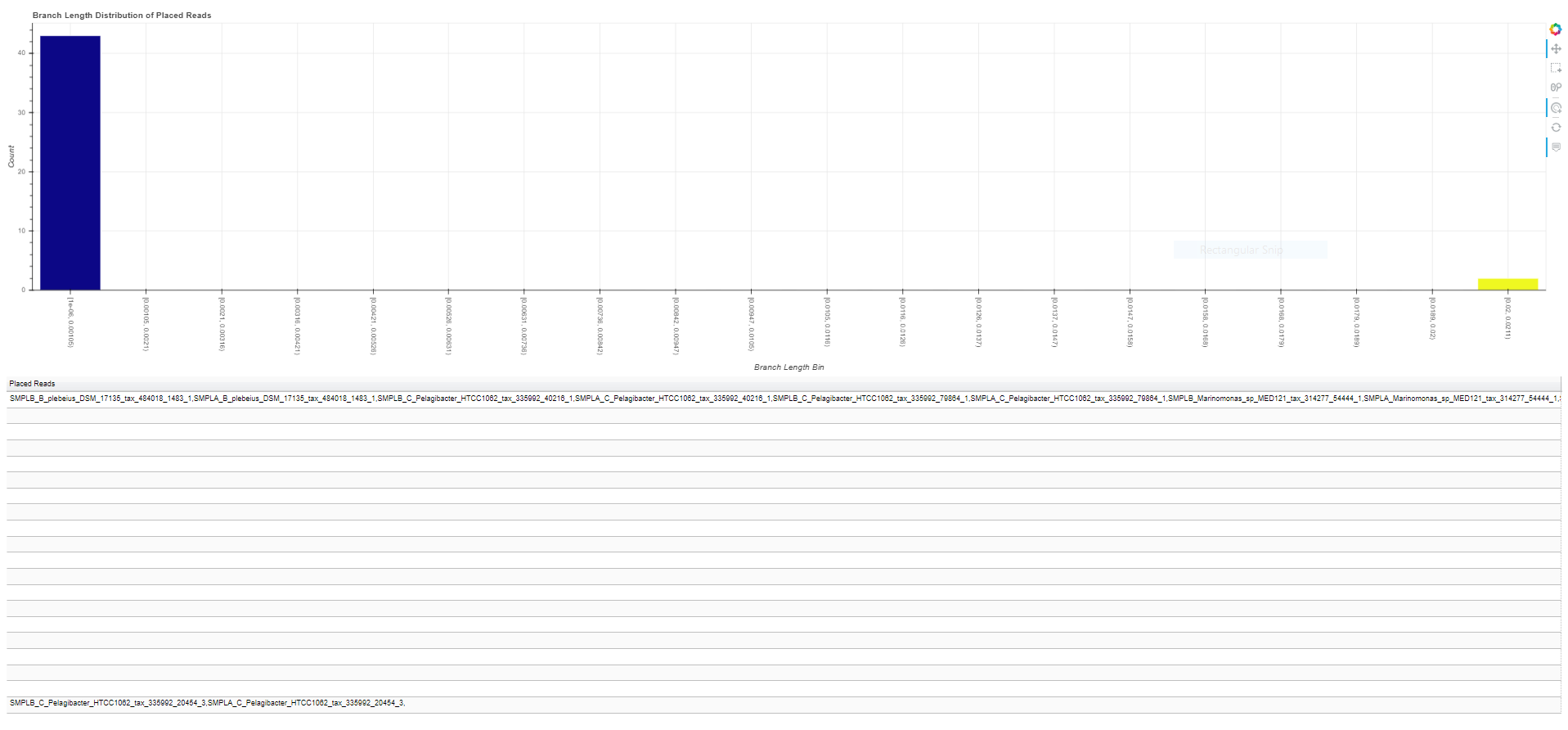
While these figures are extremely simple for this tutorial, they become more useful with complex data. For example, the following are the distributions for the more complicated dataset analyzed and described in the manuscript.
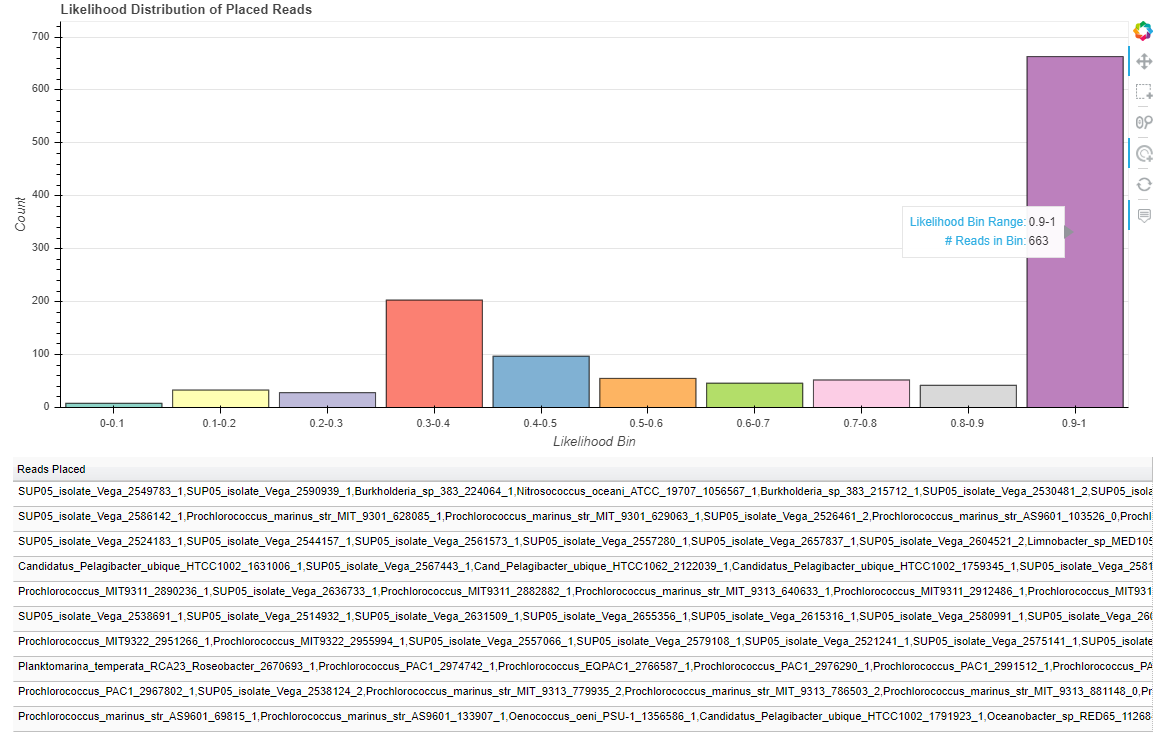
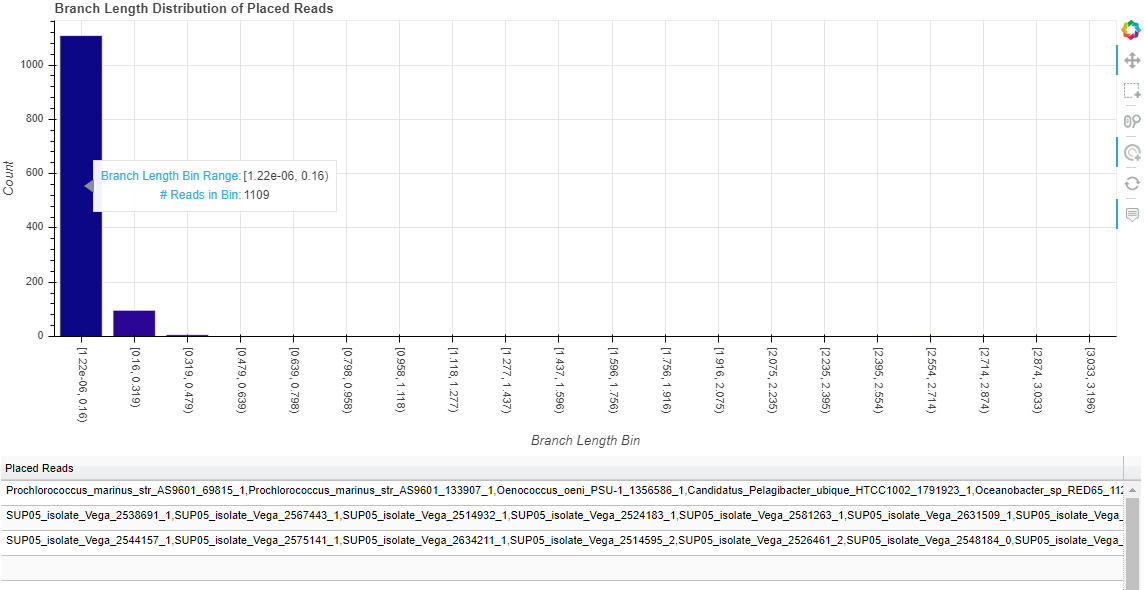
4.3: View the placements and tree with FigTree¶
The script “phan-figTreeFiles.py” will reformat the placement tree into a newick-like format that can be viewed with the phylogenetic tree viewer FigTree. A basic annotation file will also be generated that can be loaded into FigTree for additional tree visualizations (used in the color mapping of reads).
$ phan-figTreeFiles.py -i RAxML_labelledTree.rpsD_AA_placements -o rpsD
phan-figTreeFiles.py options:
| -i | Specify the tree you wish to reformat. In the example, using the labelled tree output from RAxML-EPA, but the original tree output can also be used as input. |
| -o | specify the label you would like to use in your output file names. |
You can now open the output file in the FigTree GUI tree viewer. Open the file PlacementTree_rpsD.nw with FigTree. When the “Input” window pops up, change the field to “node”.
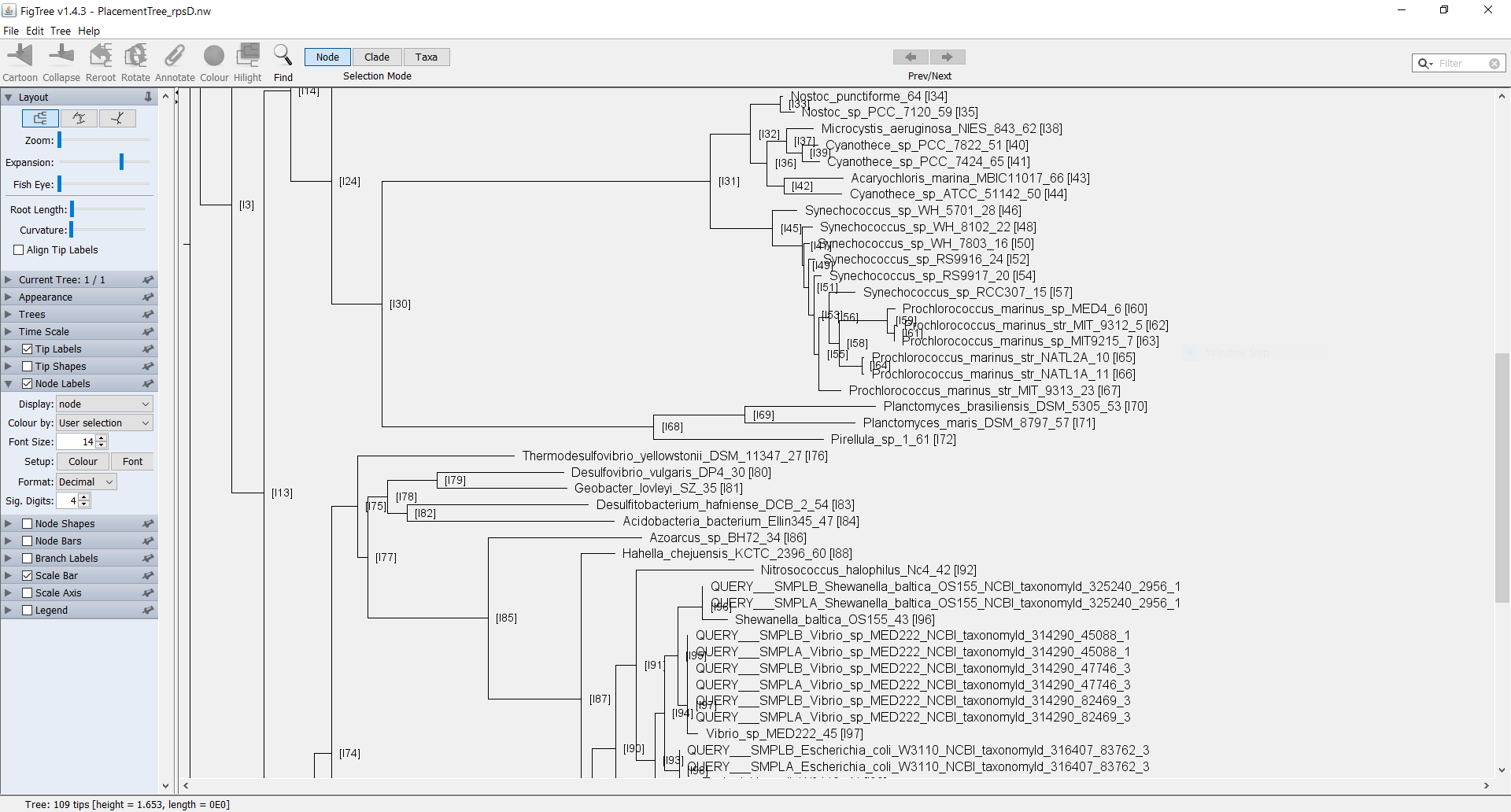
Now you can see the Tree in FigTree. Notice that the names of the terminal nodes now have the node number appended to the end of the label as [I#]. You can now add in internal node labels as well by checking the box for “Node Labels” in the left pane. If you select the arrow, the field will drop down with more details. Click on the drop-down selector next to “Display” and select the field “node”. You should now be able to see all the internal nodes labeled throughout the tree.
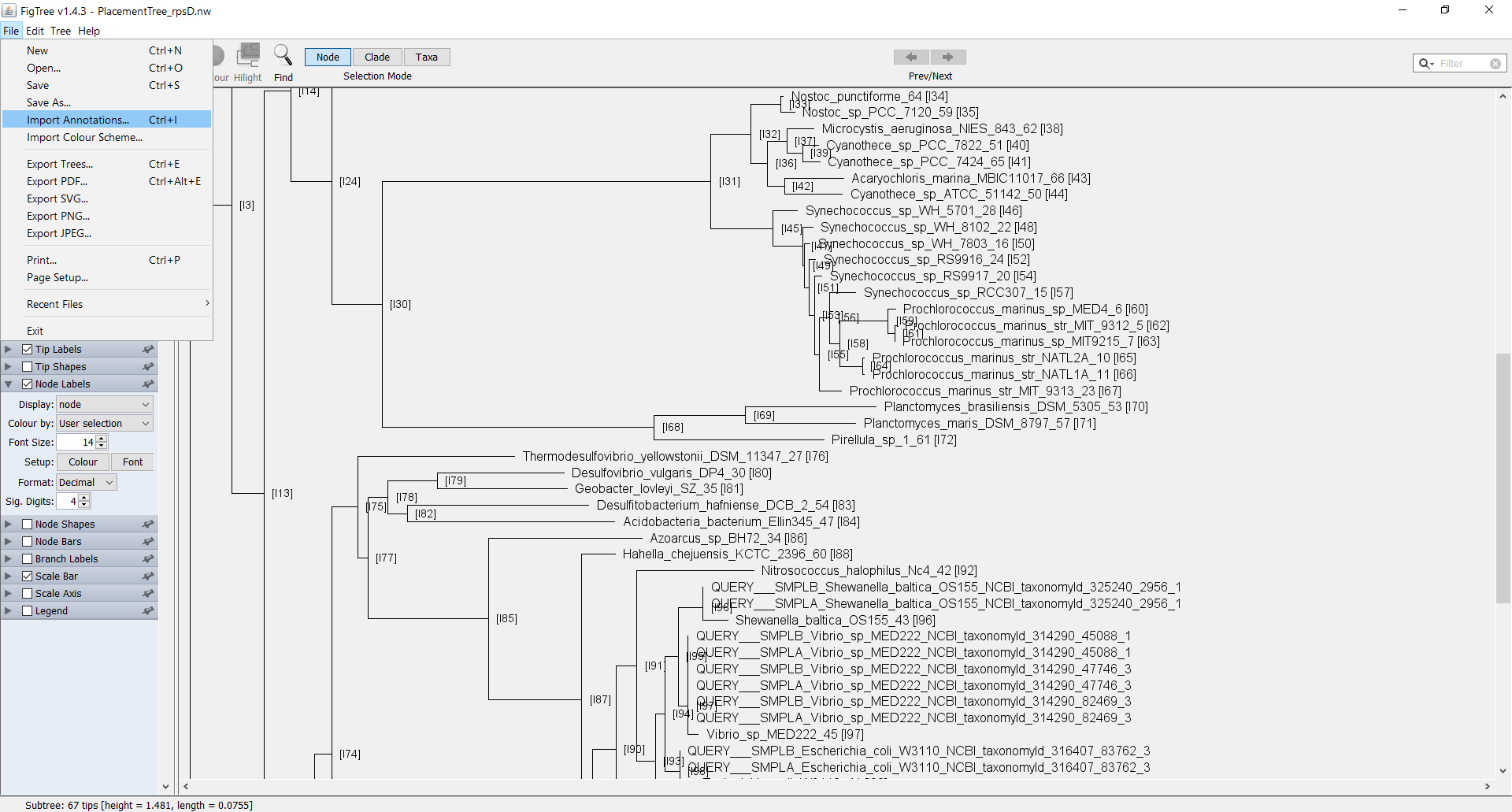
You can add further customization to the tree by using the default annotations file that is produced by phan-figTreeFiles.py called AnnotationsFile_PlacementTree_rpsD.tab. You can manually add additional labels to this file in order to add further customizations. In order to load this annotations file, in FigTree go to File > Import_Annotations and load the file AnnotationsFile_PlacementTree_rpsD.tab.
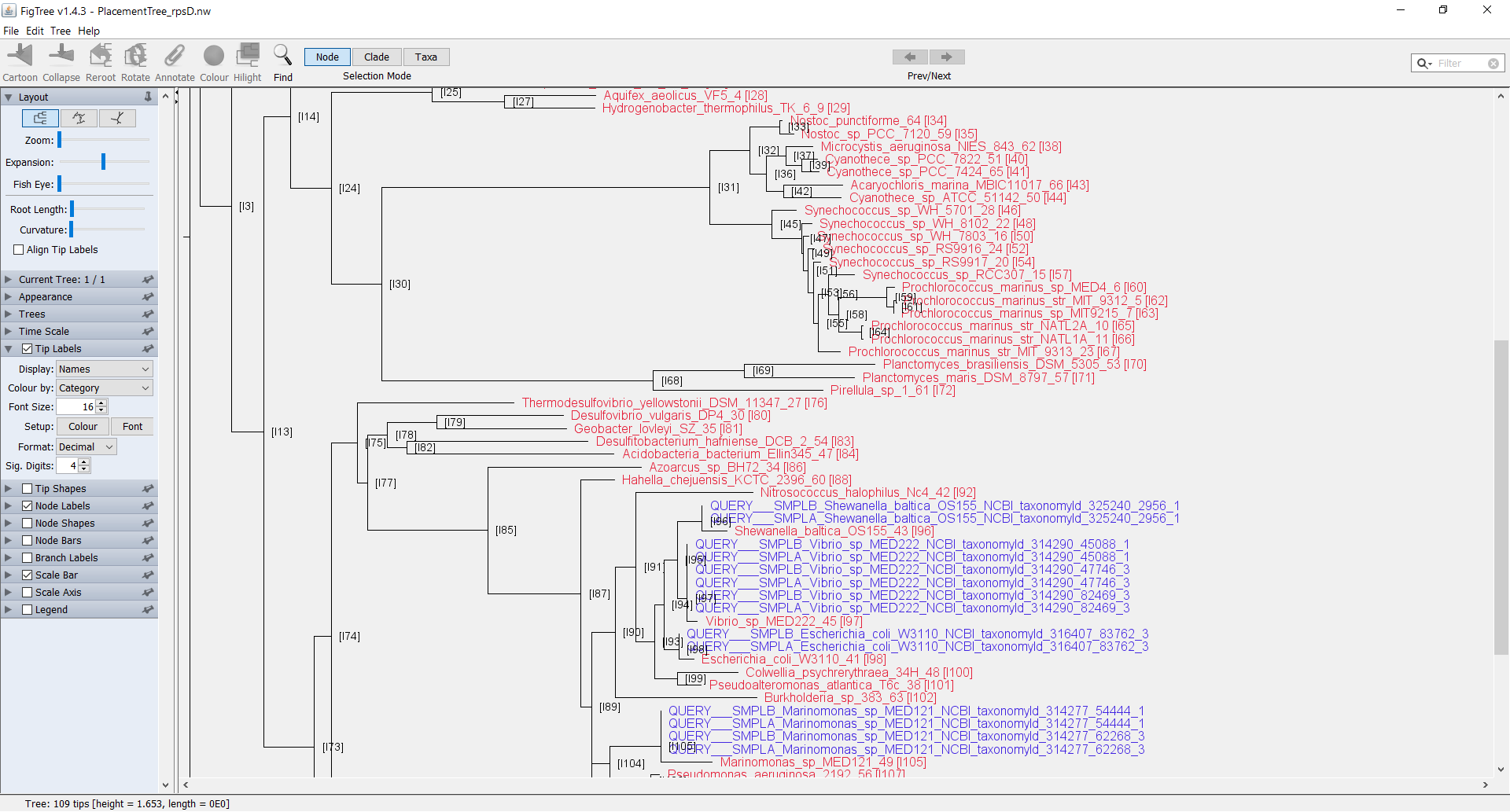
In order to visualize the annotations, select the arrow next to “Tip Labels” in the left pane. From the drop down selector next to “Colour by:” select “Category”. In order to adjust the colors further, next to “Setup:” click on the box “Colour”.
This is a very short overview on working with annotations in FigTree. Please see the FigTree documentation and discussion threads for further information.
4.4: Create annotation key for read placements¶
Generate tree file dictionary:
The RAxML-EPA output file RAxML_classification.rpsD_AA_placements classifies the placement reads by labeled node ID. In order to make the classifications more readily interpretable, a dictionary file is created based upon the tree file RAxML_originalLabelledTree.rpsD_AA_placements which acts as a key to convert labeled node number into the reference sequence name for all extant nodes.
$ phan-taxaDictionary.py -O RAxML_originalLabelledTree.rpsD_AA_placements -N rpsD_taxa_dictionary.txt
If the output is improperly named (see note about -N below) you may have to rename the output file.
$ mv rpsD_taxa_dictionary.txt_Taxa_Dictionary.txt rpsD_taxa_dictionary.txt
phan-taxaDictionary.py command line options:
| --version | Show program’s version number and exit. |
| -h, --help | Show this help message and exit. |
| -O FILE | Specify the original tree file output from RAxML-EPA that is the labeled tree by node of Ref taxa only. |
| -N STR | Specify the output file name for the taxa dictionary. Note that the -N flag may not be honored, if so you must hand rename the file. |
phan-taxaDictionary.py output file:
rpsD_taxa_dictionary.txt - The dictionary output file.
...
I6 Flavobacterium_psychrophilum_JIP0286_17
I9 Bacteroides_plebeius_DSM_17135_36
I10 Bacteroides_vulgatus_ATCC_8482_37
I8 I8
I11 Bacteroides_eggerthii_DSM_20697_32
I7 I7
I5 I5
I12 Chlorobium_limicola_DSM_245_31
...
Update tree file dictionary:
If we look at this tree, we can see that some of the internal nodes can also be relabeled from node numbers to more meaningful text labels. Add in FigTree info Some tree viewing software may require you to add the ‘.tree’ suffix to the end of the file name in order to open it. Below is a zoomed in section of the tree showing the portion of the reference tree containing *Bacteroides.

The internal nodes I8 and I7 are internal nodes containing Bacteroides. It is possible to manually update the dictionary file to reflect this, so that any placements made to these internal nodes will be labeled as Bacteroides.
Manually update rpsD_taxa_dictionary.txt:
...
I6 Flavobacterium_psychrophilum_JIP0286_17
I9 Bacteroides_plebeius_DSM_17135_36
I10 Bacteroides_vulgatus_ATCC_8482_37
I8 Bacteroides
I11 Bacteroides_eggerthii_DSM_20697_32
I7 Bacteroides
I5 I5
I12 Chlorobium_limicola_DSM_245_31
...
4.5: Reannotate read placements with meaningful labels¶
Use the dictionary file, rpsD_taxa_dictionary.txt to relabel the nodes from the RAxML-EPA classification file.
$ phan-treeLabels.py -D rpsD_taxa_dictionary.txt -C RAxML_classification.rpsD_AA_placements
phan-treeLabels.py command line options:
| --version | Show program’s version number and exit. |
| -h, --help | Show this help message and exit. |
| -D FILE | Specify the dictionary file that contains the node location ‘[I#]’ with the associated reference taxa name. |
| -C FILE | Specify the output file from RAxML-EPA that is a list of the Query sequences assigned to their locations on the reference tree |
phan-treeLabels.py output file:
RAxML_classification.rpsD_AA_placements_placement_list.csv - The reads placed by relabeled tree nodes in .csv format.
Tree_node, Read_ID
Bacteroides_plebeius_DSM_17135_36,SMPLB_Bacteroides_plebeius_DSM_17135_NCBI_taxonomyId_484018_1483_1
Shewanella_baltica_OS155_43,SMPLB_Shewanella_baltica_OS155_NCBI_taxonomyId_325240_2956_1
Bacteroides_plebeius_DSM_17135_36,SMPLA_Bacteroides_plebeius_DSM_17135_NCBI_taxonomyId_484018_1483_1
...
4.6: Tabulate reads & sort by metagenome¶
Finally, you can group and count all reads placed to unique tree labeled nodes. If nodes were labeled with the same text string (e.g. internal nodes I7 & I8 relabeled as Bacteroides, then the count will be a combination of both of these labels).
If the original BLAST database contained sequences from multiple metagenomic samples, then this script can optionally parse out the placements by metagenome. In order to utilize this feature, the reads must all be labeled with unique metagenome sample ID pre-fixes (e.g. read SMPLA_Bacteroides_plebeius_DSM_17135_NCBI_taxonomyId_484018_1483_1 is from metagenome SMPLA). A key file of the metagenomes must also be included which consists of a text file with each unique metagenomic sample ID listed per line. For the tutorial, one has been provided: Sample_list_IDs.txt
SMPLA
SMPLB
Since the tutorial consists of a database with two metagenomes, we will separate them out.
$ phan-sortPlacements.py -C RAxML_classification.rpsD_AA_placements_placement_list.csv -L Sample_list_IDs.txt
phan-sortPlacements.py command line options:
| --version | Show program’s version number and exit. |
| -h, --help | Show this help message and exit. |
| -L FILE | Specify the metagenomic sample unique identifiers - one ID per line without special characters. |
| -C FILE | Specify the reads listed according to their node which has already been relabeled by phan-taxaDict.py. |
phan-sortPlacements.py output files:
Two files are generated for every metagenomic sample. Here, we have files generate for SMPLA and SMPLB metagenomes.
RAxML_classification.rpsD_AA_placements_placement_list_SMPLA_counts.csv - A .csv file of the counts of reads by unique node label.
Tree_node, Total Reads
Escherichia_coli_W3110_41,1
Bacillus_selenitireducens_MLS10_29,2
Deinococcus_geothermalis_DSM_11300_33,3
Cand_Pelagibacter_ubique_HTCC1062_1,9
Bacteroides_plebeius_DSM_17135_36,1
Vibrio_sp_MED222_45,3
Shewanella_baltica_OS155_43,1
Marinomonas_sp_MED121_49,2
...
RAxML_classification.rpsD_AA_placements_placement_list_SMPLA_readsPlaced.csv - A .csv file of all the reads placed to each unique node label. The first entry on each line is the unique node label and all the following elements are the individual reads which placed to that node.
Tree_node, Reads_placed
Escherichia_coli_W3110_41,SMPLA_Escherichia_coli_W3110_NCBI_taxonomyId_316407_83762_3
Bacillus_selenitireducens_MLS10_29,SMPLA_Bacillus_selenitireducens_MLS10_NCBI_taxonomyId_439292_28809_3,SMPLA_Bacillus_selenitireducens_MLS10_NCBI_taxonomyId_439292_52168_3
Deinococcus_geothermalis_DSM_11300_33,SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_37835_3,SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_2 6433_3,SMPLA_Deinococcus_geothermalis_DSM_11300_NCBI_taxonomyId_319795_93937_3
...
The related files, RAxML_classification.rpsD_AA_placements_placement_list_SMPLB_counts.csv and RAxML_classification.rpsD_AA_placements_placement_list_SMPLB_readsPlaced.csv are also generated for reads from metagenomic sample SMPLB.
5: Final Summary¶
The final result of running the PHANS-C Pipeline is a list of short sequencing reads that have been characterized according to full length references. If you ran the run_tutorial.sh script, then you will find a directory called /Results which has the highlights produced from this script. In order to generate these results manually (if you followed this tutorial manually, step-by-step), run the following to generate this same directory:
$ mkdir ../Results
$ mkdir ../Results/Quantitation
$ mkdir ../Results/EPA-Out
$ mkdir ../Results/Vis-Distributions
$ cp RAxML_labelledTree.rpsD_AA_placements RAxML_originalLabelledTree.rpsD_AA_placements RAxML_classification.rpsD_AA_placements RAxML_classificationLikelihoodWeights.rpsD_AA_placements ../Results/EPA-Out/
$ cp rpsD_branchLengths_distribution_branch-lengths.html rpsD_likelihoods_distribution_likelihoodWeights.html ../Results/Vis-Distributions/
$ cp AnnotationsFile_PlacementTree_rpsD_figTree.tab PlacementTree_rpsD_figTree.nw ../Results/
$ cp RAxML_classification.rpsD_AA_placements_placement_list.csv RAxML_classification.rpsD_AA_placements_placement_list_SMPLA_counts.csv RAxML_classification.rpsD_AA_placements_placement_list_SMPLA_readsPlaced.csv RAxML_classification.rpsD_AA_placements_placement_list_SMPLB_counts.csv RAxML_classification.rpsD_AA_placements_placement_list_SMPLB_readsPlaced.csv ../Results/Quantitation/
The overall results are broken up into 3 subdirectories. /Results/EPA-Out are copies of the main output files from the EPA placement algorithm - to view the placement files in FigTree, you can use $ phan-figTreeFiles.py to generate a newick-like file that can be loaded by FigTree (See Section 4.3 for details). /Results/Vis-Distributions are copies of the interactive visualizations of the read placement metrics. And /Results/Quantitation lists copies of .csv files which list counts of reads placed to nodes on the reference tree.