Overview of PHANS-C Pipeline¶
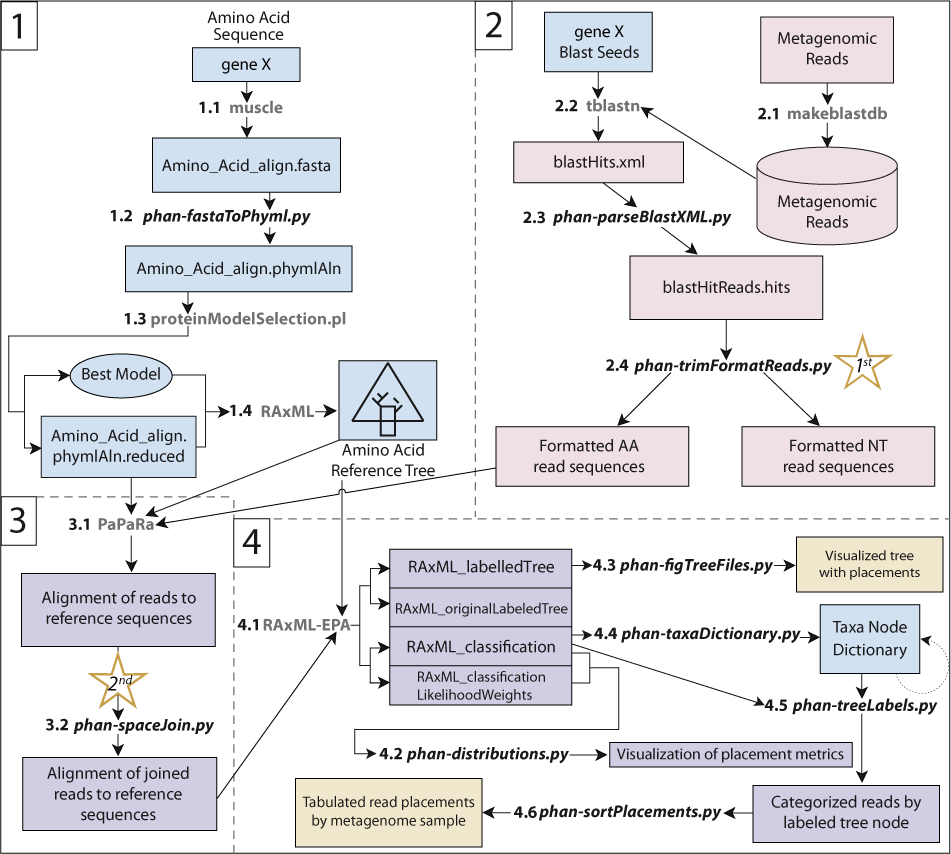
A flowchart of the bioinformatics pipeline. Objects represent files, while connecting arrows are actions on objects either through software or manual curation. Gray text between arrowed lines represents previously existing software while bold italicized black text represents interconnecting scripts described here in the PHANS-C pipeline. The PHANS-C pipeline is broken up into four major parts: 1. Full length reference sequences for the gene of interest are aligned and a reference tree is created; 2. Short reads are parsed from a large metagenomic database using local-alignment or mapping based tools. Then the recruited reads are cleaned and formatted for phylogenetic inference; 3. The unknown reads are aligned to the reference alignment; 4. Unknown aligned reads are phylogenetically placed to their least common ancestor on the reference gene tree, reads are labeled accordingly and tabulated.
Below is a table of the various programs in the PHANS-C Pipeline along with brief descriptions of the function of the program in the pipeline as well as associated citation indicating whether the program is new and specific to this work [2] or previously available academic software.
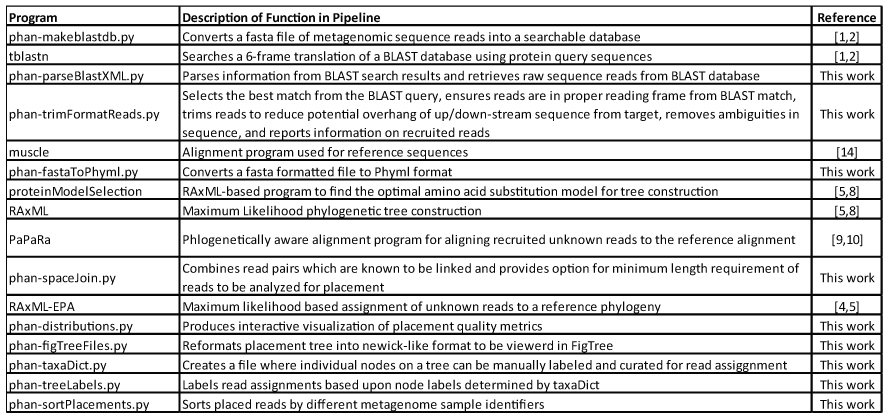
Citation numbers correspond to citations listed below.
Citations for using PHANS-C¶
Please cite the following papers if using this pipeline:
[1] MUSCLE: Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792-1797. doi: 10.1093/nar/gkh340
[2] PHANS-C Pipeline: Saunders, J. K., McKay, C., Rocap, G. (submitted). PHANS-C Pipeline: PHylogenetic Assignment of Next generation Sequences - in the Cloud.
[3] RAxML: Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30(9), 1312-1313.
[4] BLAST: Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., & Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res, 25(17), 3389-3402.
[5] PaPaRa: Berger, S. A., & Stamatakis, A. (2012). PaPaRa 2.0: a vectorized algorithm for probabilistic phylogeny-aware alignment extension. Heidelberg Institute for Theoretical Studies, https://sco.h-its.org/exelixis/pubs/Exelixis-RRDR-2012-5.pdf.
[6] RAxML Evolutionary Placement Algorithm EPA: Berger, S. A., Krompass, D. & Stamatakis, A. (2011). Performance, Accuracy, and Web Server for Evolutionary Placement of Short Sequence Reads under Maximum Likelihood. Systematic Biology (60) 291-302. doi:10.1093/sysbio/syr010
[7] Biopython: Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., … de Hoon, M. J. L. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), 1422-1423. doi: 10.1093/bioinformatics/btp163